|
|||||
|
|||||
Stress, depression and Alzheimer’s disease: The triangle of obilion?
D. FOSKOLOS*
*MD, Trainee Psychiatrist, Doctoral candidate, 2nd Neurology Department, AHEPA Hospital, Thessaloniki, Greece
This work was presented during a session entitled “Neurochemistry and dementia” in the 7th National Interdisciplinary Conference of Alzheimer’s Disease & Related Disorders, held in Thessaloniki from 16 to 20 February 2011.
Abstract
Alzheimer's disease is the most common cause of neurodegenaration and dementia among the aged population. The disease is characterized by a progressive reduction of cognitive functions, alongside other psychiatric symptoms such as psychosis and depression.
Depression is one of the most prevalent and life-threatening forms of mental illnesses and a major cause of morbidity worldwide. Estimates of prevalence range from 5 to 20% of the general population, when people with mild depressive episodes are included. Its mechanisms and pathophysiology are still not fully understood, despite the number of different theories proposed. A cognitive decline which is related to the severity of the disorder can be present among the symptoms of depression.
A number of mechanisms are considered to be implicated in the pathogenesis of depression that seems to be common in Alzheimer’s disease. At the same time there is an increasing number of clinical data, suggesting that depression in a patient’s medical record can be a risk factor for developing Alzheimer’s disease, taking into account both the number of depressive episodes, as well as the age of their onset. A possible explanation may also lie in the fact that chronic stress that is often encountered in depressed patients activates the mechanisms related to the manifestation of Alzheimer’s disease.
The purpose of this study is to attempt the amalgamation of the current body of knowledge, focusing on the correlations and common biochemical mechanisms between Alzheimer’s disease and depression, aiming to help our understanding of whether depression and chronic stress consist independent risk factors of Alzheimer’s disease. Encephalos 2011, 48(3):131-136.
Key words: Chronic stress, inflammation, pathogenesis of depression, hypothalamic-pituitary-adrenal axis, glucocorticoids.
Introduction
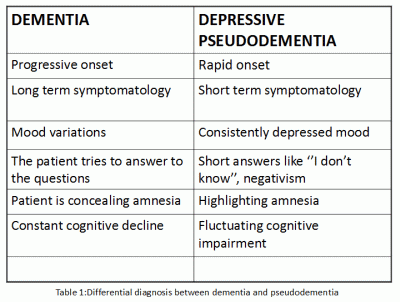
A considerable number of epidemiological studies suggest that depressive disorder in patient's history, is associated with increased risk of developing cognitive disorders1,2 and especially Alzheimer's disease.3,4 It is well known that during a depressive episode, mainly at elderly people, cognitive impairment can be present, imitating dementia. These phenomena are known as pseudodementias. The term of pseudodementia is used to describe a clinical condition characterized by depression in combination with decreased cognitive functions, which responds positively to antidepressant treatment and subside with the recession of the depression, in contrast to classical dementia which has a progressive evolution5. The concept of pseudodementia is not a subject of this work, which examines the common biochemical sites and causal associations between Alzheimer's disease and depressive disorder. Table 1 shows some feautures that help the differential diagnosis between dementia and pseudodementia.
Epidemiological data
Epidemiological studies examined the temporal relationship between one or more depressive episodes and the onset of AD, in order to understand whether depression is a prodrome symptom of AD, or an independent risk factor. The systematic meta-analysis of Ownby et al4 published in 2006, which examined the results of dozens of epidemiological studies with a total sample of 102,000 patients, concluded that: 1st: Depressive disorder in patient’s history is associated with an increased risk of developing AD. From this conclusion results that therapeutic targets might be common, since, treating effectively a depressive episode today, could probably anticipate an evolution of AD in the future.
And 2nd: There is a positive temporal correlation between the occurrence of depressive episode and the possible development of AD. In other words as the temporal distance between the depressive episode and the onset of AD gets bigger, the odds of developing AD, become more prominent. Indeed there are additional data from other studies such as that the probability of developing AD is related to the number of depressive episodes and that each new depressive episode leading to hospitalizationin in a psychiatric ward(Major Depression) increases the likelihood by 13%.2
Biochemical correlations
The attempt to connect these findings with each other and with stress occupies a significant part of recent bibliography. Experimental works6,7 as well as clinical studies8,9 indicate a strong relationship between stressful stimuli and the development of depressive disorder. This is something that we know also from our daily clinical practice, as it is understandable that someone who experiences permanent stress is more prone to depression. Recent studies highlight the causal relationships among stress and neurodegenerative diseases, particularly AD.10
Hypothalamic-pituitary-adrenal axis
A priori the mechanisms which are triggered by stressful stimuli are adaptive, facilitating or aiming to restore normal balance. These are hormonal and biochemical changes. It is known that stress triggers a neuroendocrine pathway known as the hypothalamic-pituitary-adrenal axis, or the HPA axis (Figure 1).
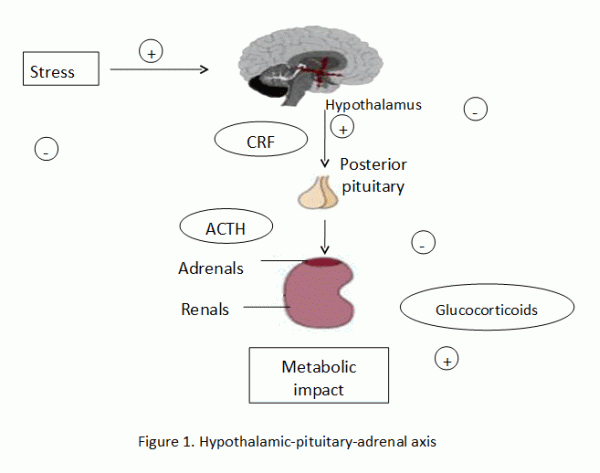
So the stressful stimuli activate the hypothalamus, which liberates the releasing factor of the corticotropin hormone known as CRF, which is acting on the pituitary gland, thus stimulating the release of adrenocorticotropic hormone or ACTH. In turn, ACTH stimulates through the blood circulation the release of glucocorticoids (GC) by the adrenal glands. Glucocorticoids (along with norepinephrine and epinephrine, released by the sympathetic nervous system) are primarily responsible for the changes occurring in the body during stressful situations in order to maximize our ability to confront the stressor that we are facing. These changes include increased cardiovascular tone, concentration of energy by the muscular system, stimulation of the immune system, inhibition of reproductive physiology and appetite, increased cerebral perfusion rates and sharpening of cognitive functions.11 The GC pass the blood-brain barrier and exercise negative retrograde regulation (negative feedback loop), to the hippocampus, hypothalamus and pituitary gland, which means that in normal conditions the excess of GC causes the halting of the HPA axis. Both in major depression as in AD, anomalies in the sequence of stress and the HPA axis are observed, thus the mechanism of negative rebound adjustment, is not working properly, an event that implies elevated levels of GC in the brain.12-16 It has been proved that the excess of these molecules in the brain plays a central pathophysiological role in both depression12, and AD,10 beeing in fact associated with a reduced activity of the GC receptors network. More specifically, a permanently elevated concentration of glucocorticoids in the bloodstream of the brain involves:
The final result is a decrease in the volume of the hippocampus and prefrontal cortex, which is found in both Alzheimer’s disease and in chronic severely depressed patients.23 The prefrontal cortex, which is offended too, is connected with the hippocampus and both structures are important for regulating mood, and memory.
The central argument of the presentation up to here, is that stress through GC, sequences a cascade of events at cellular and molecular level, which can culminate in both mood (depression) and cognitive disorders.
Inflammatory processes
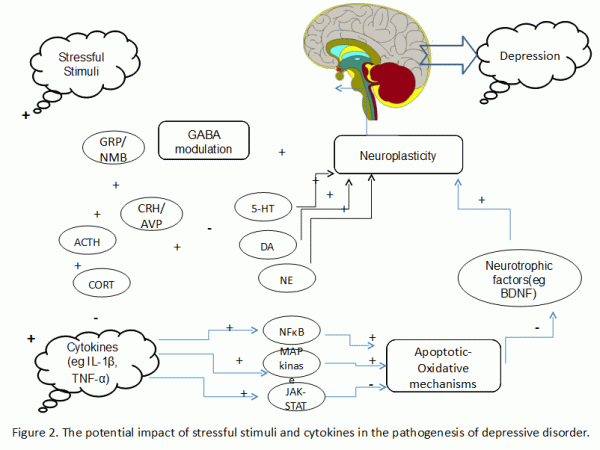
The relationship between stress and the immune system is well recognized and the idea that cytokines as factors of inflammation, may contribute to depression and neurodegeneration, is gaining ground. The involvement of the inflammatory process in Alzheimer's disease plays a central role and we should stand in the fact that some of these processes are present in depressed patients too. Some cytokines (IL-6, IL-1, TNFa), which are found to be elevated in depression, are increased also in dementia and the most important is that such inflammatory molecules appear to be involved in the pathogenesis of the AD, giving ground on the idea that both disorders can result in a sequential manner. More specifically, the activation of the immune system by an inflammatory cause, may be considered to facilitate depression, via the action of cytokines which are either entering the brain through blood circulation, or are synthesized de novo in the CNS. Those cytokines can cause processes, which are common with those activated by the classical stressful stimuli. It is even suggested that stress and cytokines can act in a synergic way24,25 (Figure 2) promoting the release of factors of the HPA axis, an event that affects serotonin, whose changes influence mood and cause depression either directly, or indirectly through the lowering of brain plasticity.
The BDN factor
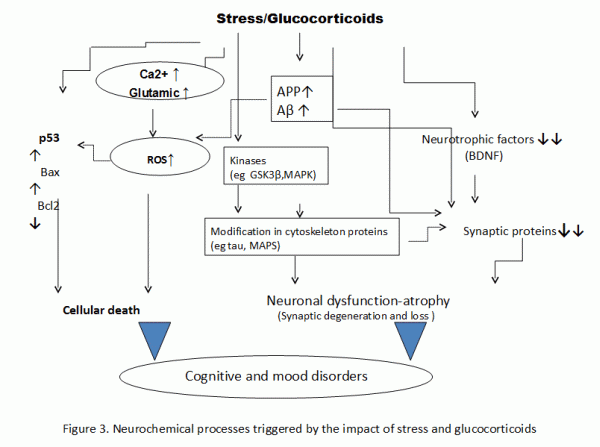
As shown in Figure 2, through the activation of other chemical agents (protein kinases and MAP), stress and cytokines could affect the evolution of apoptotic and oxidative mechanisms, wich results in the alteration of the production of the neurotrophic factor BDN (brain-derived neurotrophic factor) , in the reduction of plasticity of the brain and in depression.26 Lowered production of the BDN factor, which is observed also in AD might even arise, as shown in the 3rd picture, as a result of stress and increased levels of GC. In the same picture we see how stress and GC can trigger cell death via apoptotic processes involving proteins associated with the life cycle of the cell (P53, p21) and preapoptotic factors of the Bcl-2 family and thereby stop neurogenesis and lead to atrophy of neurons through the production of Ab from amyloid precursors and hyperphosphorylation of tau-proteins.10
Epilogue
In conclusion, it is recalled that both depression and AD, are diseases with multifactorial nature and it is clear that their etiology can not be exhausted in this work. However it seems that the effect of chronic stress by means of GC and other procedures as described above, exerts a clearly destructive action, leading to depressive disorder and AD.
REFERENCES
