|
|||
|
|||
Immunotherapy in Alzheimer's disease*
KOUTSOURAKI E.
Lecturer of 1st Dept. of Neurology, Aristotelian University, Thessaloniki, Greece
(Chairman: Professor S.J. Baloyannis)
*Oral presentation in: 5th International Congress on the Improvement of the Quality of life on Dementia, Parkinson's disease, Epilepsy, MS and Muscular disorders. 25-30 January 2007, Catania-Italy.
Encephalos 2008, 45(4):237-240.
The hallmark of immunity is specificity of recognition. In body fluids antibodies recognize small antigens which they neutralize and eliminate by various mechanisms. The discovery of the humoral immunity dates back to the 1890s, when the nobelists Emil von Behring and Shibasaburo Kitasato discovered that serum of animals immunized to corynebacterium diphtheriae could prevent diphtheria in un-immunized animals and attenuate the disease in humans. The chemical nature of the responsible serum components that neutralize and precipitate toxins as well as agglutinate and lyse bacteria was unknown at that time, but in 1907 Svante August Arrhenius undertook the first mathematical approach to disclose physical and chemicals principles of Ag-Ab interaction in a publication entitled "Immunochemistry: The application of the principles of chemistry to the study of the biological antibodies".
It was Karl Landsteiner in 1930 who provided closer insight into the specificity of Ab recognition by analyzing immunoprecipitation on a molecular level. He named it "serological reaction". Michael Heidelberger, Oswald Avery and Forrest Kendall quantified the Ag and Ab components of pneumococcus polysacharide-Ab complexes. They observed that both Ags and Abs are multivalent and that respective Abs are proteins in nature, which could exist in two forms featuring different mass and sedimentation rates (7S and 19S). These types of Abs are now called immunoglobulins (Igs) G and M (IgG and IgM).
It became apparent that Abs are glycoproteins or γ-globulins (according to their electrophoretic mobility) synthesized by mature B cells and plasma cells in response to Ag-stimulation. Their structures were elucidated by Rodney R. Porter through enzymatic cleavage by papain and by Gerald M. Edelman through reduction of interchain disulfide bonds by mercaptoethanol as being composed of two light (L) and two heavy (H) polypeptide chains in their monomeric form. In humans five Ig-classes (IgG, IgM, IgA, IgD, IgE) could be identified according to structural markers of the H chains and their multimeric assembly.
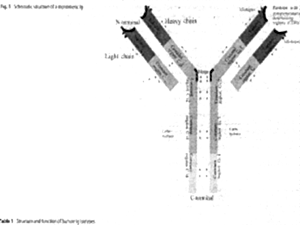
The basic structure of Igs comprises four polypeptide chains (two L and two H) connected by disulfide bridges. In humans the L chains contain one variable and one constant region whereas the H are composed of three (IgG, IgA, IgD) or four (IgM and IgE) constant regions. The N-terminal variable and constant region of one L and one H chain constitute the Fab fragment (named according to "Ag binding"), whereas the remaining two to three constant domains of the two H chains constitute the C-terminal Fc-fragment (named according to "crystalline"). IgG comprise four subclasses (IgG1, IgG2, IgG3, IgG4) and IgA two subclasses (IgA1 and IgA2).
The basic function of the Fab fragment is the binding of antigenic epitopes via complementarity determining regions (CDRs).
The principle functions of Fc are: receptor-mediated phagocytosis, cytotoxicity, release of inflammatory mediators, transport through mucosa and placenta and complement activation.
Immunoglobulins were first used for the prophylaxis and treatment of infectious diseases.After the description of a-gammaglobulinemia by Bruton in 1952 the substitution of immunoglobulins became another indication. The preparations were injected intramuscularly with the disadvantages of a very limited dose that could be given, painful irritation of the muscle, and local proteolytic degradation. An intravenous application was not possible due to the aggregates of purified immunoglobulins, which led to severe adverse reactions due to the activation of the complement cascade. The development of new purification technologies allowed the elimination of aggregates and thus preparations of immunoglobulins for intravenous use (IVIg) became available. The main indication remained substitution therapy in patients with immune defects such as a- and hypo-gammaglobulinemia.
In 1981 during the treatment of two children with hypogammaglobulinemia and coincidental idiopathic thrombocytopenic purpura it was observed increase in the platelet count after each IVIg treatment and an immunomodulatory effect of IVIg was postulated. The dose and frequency of administration (0.4 g/kg on five consecutive days) were arbitrarily chosen and proved to be efficacious in many studies.
First reports on the use of IVIg in MS were published in 1982 and 1983. In 1985 several groups reported the successful treatment of myasthenia gravis with IVIg. CIDP was the first immune-mediated neuropathy described to be responsive to IVIg followed by the Guillain Barre syndrome in 1988. Two large controlled trials in GBS in 1992 and 1997 led to the licensing of IVIg for GBS as the first neurological disease. Currently immune mediated neuropathies are probably the most common indication for IVIg in neurological diseases. However IVIg are treatment option under certain conditions in several neuroimmunological diseases.
Despite the large number of studies the precise mechanism of action of IVIg is not yet known. Immunomodulatory effects of IVIg include the inhibition of B cells, neutralization of auto-antibodies and the catabolism of antibodies. Interference with the complement system may occur at several steps in the complement cascade. T cell activation and antigen presentation can be inhibited by IVIg. Several molecular mechanisms required for the migration of leukocytes into tissue as a crucial part of an inflammatory reaction may be inhibited by IVIg. Furthermore the cellular cross-talk via the cytokine network may be modulated by IVIg. Phagocytic cells may be modulated by IVIg and inhibition of phagocytosis by monocytes and microglia has been demonstrated.
Several functions of monocytes may be modulated via Fc receptors including cytokine release and increase of antibody degradation may be blocked by IVIg rendering other IgG molecules, including autoantibodies to degradation. IVIg may also interfere with cell survival and cell death (they can protect keratinocytes, the target cell in toxic epidermal necrolysis, from apoptosis via blockade of the Fas death receptor). IVIg may inhibit TNFα mediated cytotoxicity. On the other hand, IVIg are also capable of promoting apoptosis in some lymphocytes and monocytes and terminate an immune reaction.
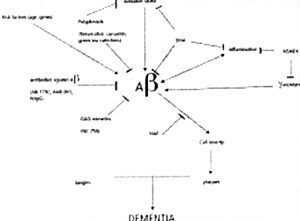
Emerging drug therapies for dementia are increasingly chosen to tackle molecular targets important in Alzheimer's disease (AD) pathobiology. Amyloid oligomers, amyloid deposits and neurofibrillary tangles (NFTs) are characteristic findings in AD. Drugs that interfere with these proteinaceous aggregates are receiving the most attention: α, β, γ- secretase modulators, inhibitors of Ab aggregation and anti-Aβ immunologic strategies. Oxidative stress and inflammatory reactions appear part of a loop of neurotoxicity, thus antioxidants and anti-inflammatory compounds have received much attention. Finally, other compounds may work by a variety of other mechanisms.
Soluble oligomers of Aβ are the earliest effectors of synaptic compromise in AD. There are 3 amyloid-based therapeutic strategies: secretase modulation, inhibition of Aβ aggregation and immunization against Aβ.
A prospective pilot study of passive immunization in AD published in J. Neurology, Neurosurgery and Psychiatry Oct;75(10):1374-5,2004 written by Dodel et al. They gave five AD patients 0.4 g/kg IVIgG on three consecutive days every four weeks for six months. Mean total Aβ concentration in the CSF decreased by 30.1% (17.3-43.5%) compared with baseline (327.5 pg/ml vs 467 pg/ml). Mean total Aβ serum concentration increased by 233% compared with baseline (558.2 pg/ml vs 240.4 pg/ml). No change was observed in CSF or serum Aβ 1-42 concentrations. ADAS-cog showed a slight improvement in the scores of four pts at six months with the fifth remaining stable. MMSE scores yielded similar findings in visual construction abilities, which often fail early in the course of AD; the MMSE score improved in three pts while remaining stable in two. No pts showed decline over the course of the study.
Evidence from longitudinal studies suggests that the mean level of decline in ADAS-cog over one year for untreated AD pts will be approximately 7-11 points and 4-6 in pts treated with cholinesterase inhibitors. No serious adverse events were reported. Three pts reported tension headaches lasting less than one day. The application of IVIg in AD pts in this setting was well tolerated. However much more investigation of IVIg and passive immunization of the treatment of AD should be done.
Limitations of the study include small size, unknown optimal dose and an unknown mechanism of action for IVIgG. If the beneficial effects of IVIgG were due to the presence of anti-Aβ antibodies it would be likely that they may have been related to a neutralizing effect on toxicity of Aβ on neurons. One could also postulate that the effects of IVIgG in the AD pts were due to altered cytokine production by microglial cells.
Ab immunization results in alleviation of memory impairment in transgenic mice. In humans, active vaccination results in a trend toward cognitive benefit in antibody responders. Surprisingly, responders show an increased rate of hippocampal atrophy by MRI. Several autopsies showed plaque clearance.
The phase II trial of vaccination with the synthetic Aβ (AN1792; AIP-001) was halted early because of encephalitis, presumably induced by T-cell immune responses, in 6% of subjects. To avoid this complication current strategies involve passive immunotherapy. Humanized monoclonal antiamyloid antibody is under development. There is evidence that anti-Aβ antibodies could bind and help clear soluble oligomers of Aβ distinct from microglial mediated clearance and peripheral sink effects, but adequately to reduce concentrations toxic to synaptic physiology. Elan's humanized monoclonal antibody (AAB-001) is designed to provide antibodies to Aβ directly to the patient, obviating the need for an immune response by the patient. It has entered phase II testing.
One may expect steady progress in various antiamyloid therapeutic strategies. It is conceivable that a number of therapies will be approved within a matter of three or five years. One of the most promising strategy is immunotherapy in AD patients.
REFERENCES
